

November 26, 2018
![]()
CDD Awarded Phase 1 SBIR Grant on Deep Learning Strategy for Drug Discovery
Burlingame, California — November 26, 2018 — Collaborative Drug Discovery, provider of CDD Vault web-based drug discovery informatics platform, announced they won a competitive, peer-reviewed Phase 1 SBIR grant from NIH NCATS, titled: “Novel Deep Learning Strategy to Better Predict Pharmacological Properties of Candidate Drugs and Focus Discovery Efforts”.
Collaborative Drug Discovery, Inc. (CDD) proposes to develop a novel approach based on deep learning neural networks to encode molecules into chemically rich vectors. We will first apply this representation to build more powerful computational models that can more accurately predict properties — such as bioactivity, ADME/ Tox, and pharmacokinetics — across libraries of molecular structures.
The ultimate goal is to leverage this representation to generate novel compounds with better combinations of properties. Both of these capabilities will help scientists to accelerate discovery of new drugs broadly across many therapeutic areas.
When building a model to predict a pharmacological property of a series of molecules, computational chemists typically start by selecting what they believe to be the relevant chemical features. Then, they assemble vectors of molecular descriptors (or fingerprints) that characterize these features in order to represent the molecules, and perform a regression analysis over the vectors.
This approach reduces dimensional complexity and makes the model tractable, but also throws away much important structural information about the molecules — a critical weakness that is widely recognized. After ~30 years of steady incremental improvements, efforts to craft better descriptors based on human intuition are unlikely to yield significant further gains.
In recent years, many research groups have tried to work around this limitation by applying deep learning (DL) techniques, but these efforts have significantly improved the prediction accuracy of computational chemistry models in only a handful of cases, where large sets of assay data are available to train the models.
In other fields, where enormous training sets are routinely available, in contrast, DL models have dramatically advanced the state of the art (e.g. image classification, voice recognition, and language translation).
Our innovative approach is to apply DL to the more focused problem of encoding the features of molecules into chemically rich vectors. We couple the encoder to a complementary decoder, creating an autoencoder, then train both neural networks jointly by asking them to try to make the output of the decoder identical to the input to the encoder.
Self-training the autoencoder in this way does not require any assay data (which are difficult to obtain at the scale that DL prefers), but instead relies only on feeding it molecular structures (which are conveniently curated in the millions).
The chemically rich vector is a narrow layer that we wedge between the encoder and the decoder to create an information bottleneck, forcing it to become rich in chemical structure information.
Our approach is to extract this chemically rich vector and repurpose it as a substitute for conventional molecular descriptors to improve existing predictive models of any type.
Like molecular descriptors, the chemically rich vectors will make existing models tractable, but without sacrificing structural detail, and the computationally intense training can be performed once, then applied to diverse problems. Preliminary tests at CDD support our hypothesis that the richer structural information preserved in these novel vectors will significantly enhance the performance and ease of use of predictive regression models.
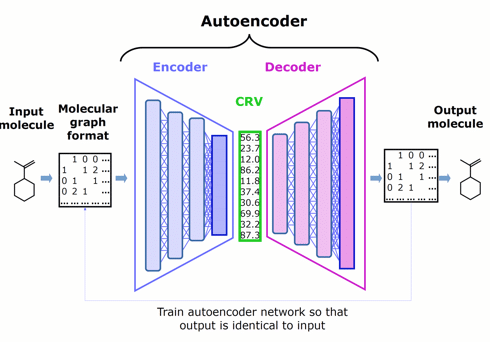 Figure 1. Schematic architecture of an autoencoder — comprised of an encoder, chemically rich vector, and decoder — that represents the starting point for Phase 1.
Figure 1. Schematic architecture of an autoencoder — comprised of an encoder, chemically rich vector, and decoder — that represents the starting point for Phase 1.
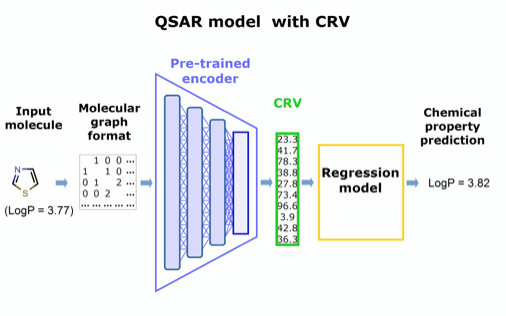 Figure 2. Concept for a QSAR model utilizing the pre-trained encoder extracted from panel in Figure 1.
Figure 2. Concept for a QSAR model utilizing the pre-trained encoder extracted from panel in Figure 1.
Computational models that predict pharmacologically relevant properties play a ubiquitous role in drug discovery research from academic laboratories to large pharmaceutical companies.
Some properties (e.g. logP) can now be modeled with such high confidence that the models have replaced the need to perform the assays, but many other critical properties (e.g. solubility, ADME, PK, hERG) remain far from this goal.
We expect that our proposed chemically rich vectors will significantly advance the state of the art beyond what can be achieved with conventional descriptors and fingerprints.
Improved models will enable researchers to select lead candidate series more effectively, explore chemical space around leads to generate novel IP more efficiently, reduce failure rates for compounds advancing through the drug discovery pipeline, and accelerate the entire drug discovery process. These benefits will be realized broadly across most therapeutic areas.
Specific Aims for Phase 1 are to:
- Re-implement the chemical autoencoder strategy summarized above with a new architecture that accepts a natural representation of the molecular graph as input, and show a substantial improvement in performance compared with our current architecture based on SMILES strings, which follow an obscure grammar.
- Exploit the chemically rich vectors to develop ~4 predictive models for diverse pharmacological properties of general interest and compare the performance of our models with the best-published benchmarks.
About this Grant
The Small Business Innovation Research (SBIR) is part of a program to enable sharing of biological data. Award Number #1R43TR002527-01 from National Center for Advancing Translational Sciences as described on NIH Reporter supports this project. This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
About Collaborative Drug Discovery, Inc.
CDD’s (www.collaborativedrug.com) flagship product, “CDD Vault®”, is used to manage chemical registration, structure-activity relationships (SAR), and securely scale collaborations. CDD Vault® is a hosted database solution for secure management and sharing of biological and chemical data. It lets you intuitively organize chemical structures and biological study data, and collaborate with internal or external partners through an easy to use web interface. Available modules within CDD Vault include Activity & Registration, Visualization, Inventory, and ELN.
A complete list of more than 60 publications and patents from CDD can be found online on our resources page at https://www.collaborativedrug.com/pages/resources.

![]()
Media Contact: Barry Bunin, Ph.D., Collaborative Drug Discovery, marketing@collaborativedrug.com.